色譜分析法、層析法,是一種分離和分析方法,現代生物企業生產過程中的核心技術之一。文字 在分析化學、有機化學、生物化學等領域有著非常廣泛的套用。色譜法利用不同物質在不同相態的選擇性分配,以流動相對固定相中的混合物進行洗脫,混合物中不同的物質會以不同的速度沿固定相移動,最終達到分離的效果。
基本介紹
- 中文名:色譜層析
- 含義:一種分離和分析方法
- 領域:化學、有機化學、生物化學
- 原理:不同物質在不同相態的選擇性分配
色譜理論,保留時間,比移值,塔板理論,Van Deemter方程,基本技術和方法,分類,柱色譜法,紙色譜法,薄層色譜法,氣液色譜法,套用,
色譜理論
保留時間
保留時間是樣品從進入色譜柱到流出色譜柱所需要的時間,不同的物質在不同的色譜柱上以不同的流動相洗脫會有不同的保留時間,因此保留時間是色譜分析法比較重要的參數之一。
保留時間由物質在色譜中的分配係數決定:
tR = t0(1 + KVs / Vm)
式中tR表示某物質的保留時間,t0是色譜系統的死時間,即流動相進入色譜柱到流出色譜柱的時間,這個時間由色譜柱的孔隙、流動相的流速等因素決定。K為分配係數,VsVm表示固定相和流動相的體積。這個公式又叫做色譜過程方程,是色譜學最基本的公式之一。
比移值
在薄層色譜中沒有樣品進入和流出固定相的過程,因此人們用比移值標示物質的色譜行為。比移值是一個與保留時間相對應的概念,它是樣品點在色譜過程中移動的距離與流動相前沿移動距離的比值。與保留時間一樣,比移值也由物質在色譜中的分配係數決定:
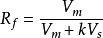
其中Rf是比移值,K表示色譜分配係數,VsVm表示固定相和流動相的體積。
塔板理論
基於熱力學的塔板理論。塔板理論是色譜學的基礎理論,塔板理論將色譜柱看作一個分餾塔,待分離組分在分餾塔的塔板間移動,在每一個塔板內組分分子在固定相和流動相之間形成平衡,隨著流動相的流動,組分分子不斷從一個塔板移動到下一個塔板,並不斷形成新的平衡。一個色譜柱的塔板數越多,則其分離效果就越好。
根據塔板理論,待分離組分流出色譜柱時的濃度沿時間呈現二項式分布,當色譜柱的塔板數很高的時候,二項式分布趨於常態分配。則流出曲線上組分濃度與時間的關係可以表示為:

這一方程稱作流出曲線方程,式中Ct為t時刻的組分濃度;C0為組分總濃度,即峰面積;σ為半峰寬,即常態分配的標準差;tR為組分的保留時間。
根據流出曲線方程人們定義色譜柱的理論塔板高度為單位柱長度的色譜峰方差:

理論塔板高度越低,在單位長度色譜柱中就有越高的塔板數,則分離效果就越好。決定理論塔板高度的因素有:固定相的材質、色譜柱的均勻程度、流動相的理化性質以及流動相的流速等。
塔板理論是基於熱力學近似的理論,在真實的色譜柱中並不存在一片片相互隔離的塔板,也不能完全滿足塔板理論的前提假設。如塔板理論認為物質組分能夠迅速在流動相和固定相之間建立平衡,還認為物質組分在沿色譜柱前進時沒有徑向擴散,這些都是不符合色譜柱實際情況的,因此塔板理論雖然能很好地解釋色譜峰的峰型、峰高,客觀地評價色譜柱地柱效,卻不能很好地解釋與動力學過程相關的一些現象,如色譜峰峰型的變形、理論塔板數與流動相流速的關係等。
Van Deemter方程
基於動力學的Van Deemter方程。Van Deemter方程是對塔板理論的修正,用於解釋色譜峰擴張和柱效降低的原因。塔板理論從熱力學出發,引入了一些並不符合實際情況的假設,Van Deemter方程則建立了一套經驗方程來修正塔板理論的誤差。
Van Deemter方程將峰形的改變歸結為理論塔板高度的變化,理論塔板高度的變化則源於若干原因,包括渦流擴散、縱向擴散和傳質阻抗等。
由於色譜柱內固定相填充的不均勻性,同一個組分會沿著不同的路徑通過色譜柱,從而造成峰的擴張和柱效的降低。這稱作渦流擴散
縱向擴散是由濃度梯度引起的,組分集中在色譜柱的某個區域會在濃度梯度的驅動下沿著徑向發生擴散,使得峰形變寬柱效下降。
傳質阻抗本質上是由達到分配平衡的速率帶來的影響。實際體系中,組分分子在固定相和流動相之間達到平衡需要進行分子的吸附、脫附、溶解、擴散等過程,這種過程稱為傳質過程,阻礙這種過程的因素叫做傳質阻抗。在理想狀態中,色譜柱的傳質阻抗為零,則組分分子流動相和固定相之間會迅速達到平衡。在實際體系中傳質阻抗不為零,這導致色譜峰擴散,柱效下降。
在氣相色譜中Van Deemter方程形式為:

其中H為塔板數,A為渦流擴散係數,B為縱向擴散係數,C為傳質阻抗係數,μ為流動相流速。
在高效液相色譜中,由於流動相粘度遠遠高於氣相色譜,縱向擴散對峰型的影響很小,可以忽略不計算,因而Van Deemter方程的形式為:
H = A + Cμ
基本技術和方法
分類
吸附色譜是利用吸附劑對被分離物質的吸附能力不同,用溶劑或氣體洗脫,以使組分分離。常用的吸附劑有氧化鋁、矽膠、聚醯胺等有吸附活性的物質。
分配色譜是利用溶液中被分離物質在兩相中分配係數不同,以使組分分離。其中一相為液體,塗布或使之鍵合在固體載體上,稱固定相;另一相為液體或氣體,稱流動相。常用的載體有矽膠、硅藻土、矽鎂型吸附劑與纖維素粉等。
離子交換色譜是利用被分離物質在離子交換樹脂上的離子交換勢不同而使組分分離。常用的有不同強度的陽、陰離子交換樹脂,流動相一般為水或含有有機溶劑的緩衝液。
排阻色譜又稱凝膠色譜或凝膠滲透色譜,是利用被分離物質分子量大小的不同和在填料上滲透程度的不同,以使組分分離。常用的填料有分子篩、葡聚糖凝膠、微孔聚合物、微孔矽膠或玻璃珠等,可根據載體和試樣的性質,選用水或有機溶劑為流動相。
分離後各成分的檢出,應採用各單體中規定的方法。通常用柱色譜、紙色譜或薄層色譜分離有色物質時,可根據其色帶進行區分,對有些無色物質,可在245-365nm的紫外燈下檢視。紙色譜或薄層色譜也可噴顯色劑使之顯色。薄層色譜還可用加有螢光物質的薄層矽膠,採用螢光熄滅法檢視。用紙色譜進行定量測定時,可將色譜斑點部分剪下或挖取,用溶劑溶出該成分,再用分光光度法或比色法測定,也可用色譜掃瞄器直接在紙或薄層板上測出,也可用色譜掃瞄器直接以紙或薄層板上測出。柱色譜、氣相色譜和高效液相色譜可用接於色譜柱出口處的各種檢測器檢測。柱色譜還可分部收集流出液後用適宜方法測定。
柱色譜法
【柱色譜法】( Column chromatography)
所用色譜管為內徑均勻、下端縮口的硬質玻璃管,下端用棉花或玻璃纖維塞住,管內裝有吸附劑。色譜柱的大小,吸附劑的品種和用量,以及洗脫時的流速,均按各單體中的規定。吸附劑的顆粒應儘可能保持大小均勻,以保證良好的分離效果,除另有規定外通常多採用直徑為0.07-0.15mm的顆粒。吸附劑的活性或吸附力對分離效果有影響,應予注意。
吸附劑的填裝 乾法:將吸附劑一次加入色譜管,振動管壁使其均勻下沉,然後沿管壁緩緩加入開始層析時使用的流動相,或將色譜管下端出口加活塞,加入適量的流動相,旋開活使流動相緩緩滴出,然後自管頂緩緩加入吸附劑,使其均勻地潤濕下沉,在管內形成鬆緊適度的吸附層。操作過程中應保持有充分的流動相留在吸附層的上面。濕法:將吸附劑與流動相混合,攪拌以除去空氣泡,徐徐傾入色譜管中,然後再加入流動相,將附著於管壁的吸附劑洗下,使色譜柱表面平整。
俟填裝吸附劑所用流動相從色譜柱自然流下,液面將柱表面相平時,即加試樣溶液。
試樣的加入 除另有規定外,將試樣溶於層析時使用的流動相中,再沿色譜管壁緩緩加入。注意勿使吸附劑翻起。或將試樣溶於適當的溶劑中。與少量吸附劑混勻,再使溶劑揮發去盡後使呈鬆散狀;將混有試樣的吸附劑加在已製備好的色譜柱上面。如試樣在常用溶劑中不溶解,可將試樣與適量的吸附劑在乳缽中研磨混勻後加入。
洗脫 除另有規定外,通常按流動相洗脫能力大小,遞增變換流動相的品種和比例,分別分部收集流出液,至流出液中所含成分顯著減少或不再含有時,再改變流動相的品種和比例。操作過程中應保持有充分的流動相留在吸附層的上面。
紙色譜法
【紙色譜法】(Paper chromatography)
以紙為載體,用單一溶劑或混合溶劑進行分配。亦即以紙上所含水分或其他物質為固定相,用流動相進行展開的分配色譜法。
所用濾紙應質地均勻平整,具有一定機械強度,必須不含會影響色譜效果的雜質,也不應與所用顯色劑起作用,以免影響分離和鑑別效果,必要時可作特殊處理後再用。
試樣經層析後可用比移值(Rf)表示各組成成分的位置(比移值=原點中心至色譜斑點中心的距離與原點中心至流動相前沿的距離之比),由於影響比移值的因素較多,因此一般採用在相同實驗條件下對照物質對比以確定其異同。作為單體鑑別時,試樣所顯主色譜斑點的顏色(或螢光)與供置,應與對照(標準)樣所顯主色的譜斑點或供試品-對照品(1∶1)混合所顯的主色譜斑點相同。作為質量指標(純度)檢查時,可取一定量的試樣,經展開後,按各單體的規定,檢視其所顯雜質色譜斑點的個數或呈色(或螢光)的強度。作為含量測定時,可將色譜斑點剪下洗脫後,再用適宜的方法測定,也可用色譜掃瞄器測定。
1、下行法 所用色譜缸一般為圓形或長方形玻璃缸,缸上有磨口玻璃蓋,應能密閉,蓋上有孔,可插入分液漏斗,以加入流動相。在近缸頂端有一用支架架起的玻璃槽作為流動相的容器,槽內有一玻璃棒,用以支持色譜濾紙使其自然下垂,避免流動相沿濾紙與溶劑槽之間發生虹吸現象。
取適當的色譜濾紙按纖維長絲方向切成適當大小的紙條,離紙條上端適當的距離(使色譜紙上端能足夠浸入溶劑槽內的流動相中,並使點樣基線能在溶劑槽側的玻璃支持棒下數厘米處)用鉛筆劃一點樣基線,必要時色譜紙下端可切成鋸齒形,以便於流動相滴下。
將試樣溶於適當的溶劑中,製成一定濃度的溶劑。用微量吸管或微量注射器吸取溶劑,點於點樣基線上,溶液宜分次點加,每次點加後,俟其自然乾燥、低溫烘乾或經溫熱氣流吹乾。樣點直徑一般不超過0.5cm,樣點通常應為圓形。
將點樣後的色譜濾紙上端放在溶劑槽內,並用玻璃棒壓住,使色譜紙通過槽側玻璃支持棒自然下垂,點樣基線在支持棒下數厘米處。色譜開始前,色譜缸內用各單體中所規定的溶劑的蒸氣飽和,一般可在色譜缸底部放一裝有流動相的平皿,或將浸有流動相的濾紙條附著在色譜缸的內壁上,放置一定時間,俟溶劑揮發使缸內充滿飽和蒸氣。然後添加流動相,使浸沒溶劑槽內濾紙,流動相即經毛細管作用沿濾紙移動進行展開至規定距離後,取出濾紙,標明流動相前沿位置,俟流動相揮散後按規定方法檢出色譜斑點。
2、上行法 色譜缸基本和下行法相似,唯除去溶劑槽和支架,並在色譜缸蓋上的孔中加塞,塞中插入玻璃懸鉤,以便將點樣後的色譜濾紙掛在鉤上。色譜濾紙一般長約25cm,寬度則視需要而定。必要時可將色譜濾紙捲成筒形。點樣基線距底邊約2.5cm,點樣方法與下行法相同。色譜缸內加入適量流動相,放置,俟流動相蒸氣飽和後,再下降懸鉤,使色譜濾紙浸入流動相約0.5cm,流動相即經毛細管作用沿色譜濾紙上升,除另有規定外,一般展開至15cm後,取出晾乾,按規定方法檢視。
色譜可以向一個方向進行,即單向色譜;也可進行雙向色譜,即先向一個方向展開,取出,俟流動相完全揮發後,將濾紙轉90°,再用原流動相或另一種流動相進行展。亦可多次展開,連續展或徑向色譜等。
薄層色譜法
【薄層色譜法】(Thin-layer chromatography)
按各單體所規定的載體,放入適當容器,加入適量水以配成懸浮液,在厚度均勻一致的50×200mm或200×200mm平滑玻璃板上將此懸浮液均布成0.25mm的厚度,風乾後一般在110℃下乾燥0.5-1h(或按單體規定)。
以離薄層板一端約25mm的位置作為點樣基線,用微量吸管按規定量吸取試樣液和對照(標準)液,點於基線上,點與點之間的距離在10mm以上,液點的直徑約3mm,風乾後,基線一端向下,將薄層板放入展開溶劑,溶劑層深10mm,並預經開展溶劑的蒸汽飽和。在展開溶劑從基線上升至規定距離(一般為15cm)後,取出薄層板,風乾,然後按規定的方法,對斑點的位置和顏色進行檢查。
【氣相色譜法】(Gas chromatography)
在對裝置進行調試後,按各單體的規定條件調整柱管、檢測器、溫度和載氣流量。進樣口溫度一般應高於柱溫30-50度。如用火焰電離檢測器,其溫度應等於或高於柱溫,但不得低於100℃,以免水汽凝結。色譜上分析成分的峰的位置,以滯留時間(從注入試樣液到出現成分最高峰的時間)和滯留容量(滯留時間×載氣流量)來表示。這些在一定條件下,就能反應出物質所具有特殊值,並據此確定試樣成分。
根據色譜上出現的物質成分的峰面積或峰高進行定量。峰面積可用面積測定儀測定,按半寬度法求得(即以峰1/2處的峰寬×峰高求得)。峰高的測定方法是從峰高的頂點向記錄紙橫座標準垂線,找出此垂線與峰的兩下端聯結線的交點,即以此交點至峰頂點的距離長度為峰高。
定量方法可分以下三種:
1、內標準法 取標準被測成分,按依次增加或減少的已知階段量,各自分別加入各單體所規定的定量內標準物質中,調製標準溶液。分別取此標準液的一定量注入色譜柱,根據色譜圖取標準被測成分的峰面積和峰高和內標物質的峰面積和峰高的比例為縱坐標,取標準被測成分量和內標物質量之比,或標準被測成分量為橫坐標,製成標準曲線。
然後按單體中所規定的方法調製試樣液。在調製試樣液時,預先加入與調製標準液時等量的內標物質。然後按製作標準曲線時的同樣條件下得出的色譜,求出被測成分的峰面積或峰高和內標物質的峰積或峰高之比,再按標準曲線求出被測成分的含量。
所用的內標物質,應採用其峰面積的位置與被測成分的峰的位置儘可能接近並與被測成分以外的峰位置完全分離的穩定的物質。
2、絕對標準曲線法 取標準被測成分 按依次增加或減少階段法,各自調製成標準液,注入一定量後,按色譜圖取標準被測成分的峰面積或峰高為縱坐標,而以標準被測成分的含量為橫坐標,製成標準曲線。然後按單體中所規定的方法製備試樣液。取試樣液按制標準曲線時相同的條件作出色譜,求出被測成分的峰面積和峰高,再按標準曲線求出被測成分的含量。
3、峰面積百分率法 以色譜中所得各種成分的峰面積的總和為100,按各成分的峰面積總和之比,求出各成分的組成比率。
氣液色譜法
【氣液色譜法】(gas-liquid chromatography)
這時所指的氣液色譜法,主要用於各種香料物質的分析,基本條件和參數主要依照美國精油協會(EOA)於1979年所建議的方法。其基本原理、操作、標準狀態等均與上述氣相色譜法相同。
1、柱 用304號合金所制不鏽鋼管,長3m,內徑2.16-2.57mm,外徑3.18mm。底物:極性柱為聚乙二醇20M(Carbowax 20M),分子量約2萬;非極性柱為氣相色譜級甲基矽氧烷(SE-30),或二甲基矽氧烷(OV-1或OV-101)。底物濃度:重量的105。固體載體:10目或20目熔融煅燒過的硅藻土,經矽烷化和酸洗後,其自由傾落密度為0.2g/cm3,最小120目,最大80目。裝填密度每cm3應大於0.24g。
【分析狀態】
極性柱:起始溫度,最低75度;最終溫度,最高225度。升溫速度,每分鐘2-8度。
非極性柱:起始溫度,最低75度;最終溫度,不超過275度;升溫速度,每分鐘2-8度。
進樣溫度:225-250度。試樣量:0.1-1μl。
檢測器:用熱導池。檢測器的操作條件應維持恆定。
套用
色譜法的套用可以根據目的分為製備性色譜和分析性色譜兩大類。
製備性色譜的目的是分離混合物,獲得一定數量的純淨組分,這包括對有機合成產物的純化、天然產物的分離純化以及去離子水的製備等。相對於色譜法出現之前的純化分離技術如重結晶,色譜法能夠在一步操作之內完成對混合物的分離,但是色譜法分離純化的產量有限,只適合於實驗室套用。
分析性色譜的目的是定量或者定性測定混合物中各組分的性質和含量。定性的分析性色譜有薄層色譜、紙色譜等,定量的分析性色譜有氣相色譜、高效液相色譜等。色譜法套用於分析領域使得分離和測定的過程合二為一,降低了混合物分析的難度縮短了分析的周期,是目前比較主流的分析方法。在中華人民共和國藥典中,共有超過600種化學合成藥和超過400種中藥的質量控制套用了高效液相色譜(High performance liquid chromatography)的方法。
