多體攝動理論即多體微擾理論,是一種基於分子軌道理論的高級量子化學計算方法。這種方法以Hartree-Fock方程的自洽場解為基礎,套用微擾理論,獲得考慮了相關能的多電子體系近似解,其計算精度與組態相互作用方法的DCI接近,但計算量遠小於DCI,是套用比較廣泛的高級量子化學計算方法。
基本介紹
- 中文名:多體攝動理論
- 外文名:Møller–Plesset perturbation theory
- 別稱:多體微擾理論
歷史,基本假設,能量和波函式的各級近似,零級能量和波函式,一級校正,二級校正,高級校正,方法評價,參見,
歷史
多體微擾理論是由量子化學家Møller和Plesset在1934年提出的,所以這一方法也經常以二人的名字所寫MP表示,MPn表示的是多體微擾n級近似。
基本假設
多體微擾理論是以Hartree-Fock方程為基礎的,套用微擾法處理的計算方法。微擾法要求將複雜體系的哈密頓運算元分解為可精確求解項和微擾項兩部分,在多體微擾理論中,引入Hartree-Fock哈密頓運算元的概念:



將多電子體系哈密頓運算元分解為Hartree-Fock哈密頓運算元和微擾項的代數和:



能量和波函式的各級近似
零級能量和波函式
在多體微擾理論下,基態零級能量就是構成基態斯萊特行列式的各分子軌道軌道能的代數和,零級波函式就是基態斯萊特行列式波函式。可以看出,多體微擾理論的零級能量精度甚至不如Hartree-Fock方程所得的能量。
一級校正
根據微擾理論,能量的一級校正 為:
為:





二級校正
可以看到,經過能量的一級校正後體系能量為:
根據微擾理論,體系基態能量的二級校正 為:
為:



可以證明,只有對雙激發的斯萊特行列式才有 ,所以體系能量的二級校正為:
,所以體系能量的二級校正為:

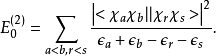
最終體系經過二級校正的基態能量為:




考慮二級校正的多體微擾計算簡稱MP2。
高級校正
更高級的校正是以較低級校正為計算基礎的,隨著校正級別的提高,計算量也急劇增加,理論上講,隨著校正級別的提高最終的體系能量會逐漸逼近真實值。目前的計算方法最高可以進行MP5計算,即體系能量的五級校正。
方法評價
多體微擾理論方法是一種量子化學高級計算方法,在考慮相關能的計算方法中,多體微擾理論方法是計算量相對最小的。MP1可以達到HF方程的計算精度,MP2一般可以達到60%的相關能,與DCI方法相當,但計算過程僅需要計算少量雙電子積分,遠遠小於DCI;MP4一般可以達到85%的相關能。
MPn方法是一個大小一致的方法,即對電子數不同的體系,使用MPn計算的精度是相同的,這一特性使得MPn方法特別適合進行化學反應的模擬計算。但是由於MPn方法以HF方程為基礎,因而受到HF方程的局限,對於那些套用HF方程不能很好處理的體系,如非限制性開殼層體系,MPn方法也不能很好處理。
參見
- Hartree-Fock方程
